Getting started¶
Best practices are implemented in a python framework.
clustering of small RNA sequences¶
seqcluster generates a list of clusters of small RNA sequences, their genome location, their annotation and the abundance in all the sample of the project
REMOVE ADAPTER
I am currently using cutadapt:
cutadapt --adapter=$ADAPTER --minimum-length=8 --untrimmed-output=sample1_notfound.fastq -o sample1_clean.fastq -m 17 --overlap=8 sample1.fastq
COLLAPSE READS
To reduce computational time, I recommend to collapse sequences, also it would help to apply filters based on abundances. Like removing sequences that appear only once.
seqcluster collapse -f sample1_clean.fastq -o collapse
Here I am only using sequences that had the adapter, meaning that for sure are small fragments.
PREPARE SAMPLES
seqcluster prepare -c file_w_samples -o res --minl 17 --minc 2 --maxl 45
the file_w_samples should have the following format:
lane1_sequence.txt_1_1_phred.fastq cc1
lane1_sequence.txt_2_1_phred.fastq cc2
lane2_sequence.txt_1_1_phred.fastq cc3
lane2_sequence.txt_2_1_phred.fastq cc4
two columns file, where the first column is the name of the file with the small RNA sequences for each sample, and the second column in the name of the sample.
The fastq files should be like this:
@seq_1_x11
CCCCGTTCCCCCCTCCTCC
+
QUALITY_LINE
@seq_2_x20
TGCGCAGTGGCAGTATCGTAGCCAATG
+
QUALITY_LINE
</pre>
Where _x[09] indicate the abundance of that sequence, and the middle number is the index of the sequence.
This script will generate: seqs.fastq and seqs.ma.
* seqs.fastq: have unique sequences and unique ids
* seqs.ma: is the abundance matrix of all unique sequences in all samples
ALIGNMENT
You should use an aligner to map seqs.fa to your genome. A possibility is bowtie or STAR. From here, we need a file in BAM format for the next step. VERY IMPORTANT: the BAM file should be sorted
bowtie -a --best --strata -m 5000 INDEX seqs.fastq -S | samtools view -Sbh /dev/stdin | samtools sort -o /dev/stdout temp > seqs.sort.bam
or
STAR --genomeDir $star_index_folder --readFilesIn res/seqs.fastq --alignIntronMax 1 --outFilterMultimapNmax 1000 --outSAMattributes NH HI NM --outSAMtype BAM SortedByCoordinate
CLUSTERING
seqcluster cluster -a res/Aligned.sortedByCoord.out.bam -m res/seqs.ma -g $GTF_FILE -o res/cluster -ref PATH_TO_GENOME_FASTA --db example
- -a is the SAM file generated after mapped with your tool, which input has been seqs.fa
- -m the previous seqs.fa
- -b annotation files in bed format (see below examples) [deprecated]
- -g annotation files in gtf format (see below examples) [recommended]
- -i genome fasta file used in the mapping step (only needed if -s active)
- -o output folder
- -ref genome fasta file. Needs
faifile as well there. (i.ehg19.fa,hg19.fa.fai) - -d create debug logging
- -s construction of putative precursor (NOT YET IMPLEMENTED)
- –db (optional) will create sqlite3 database with results that will be used to browse data with html web page (under development)
Example of a bed file for annotation (the fourth column should be the name of the feature):
chr1 157783 157886 snRNA 0 -
Strongly recommend gtf format. Bed annotation is deprecated. Go here to know how to download data from hg19 and mm10.
Example of a gtf file for annotation (the third column should be the name of the feature and the value after gene name attribute is the specific annotation):
chr1 source miRNA 1 11503 . + . gene name 'mir-102' ;
hint: scripts to generate human and mouse annotation are inside seqcluster/scripts folder.
OUTPUTS
counts.tsv: count matrix that can be input of downstream analysessize_counts.tsv: size distribution of the small RNA by annotation groupseqcluster.json: json file containing all informationlog/run.log: all messages at debug levellog/trace.log: to keep trace of algorithm decisions
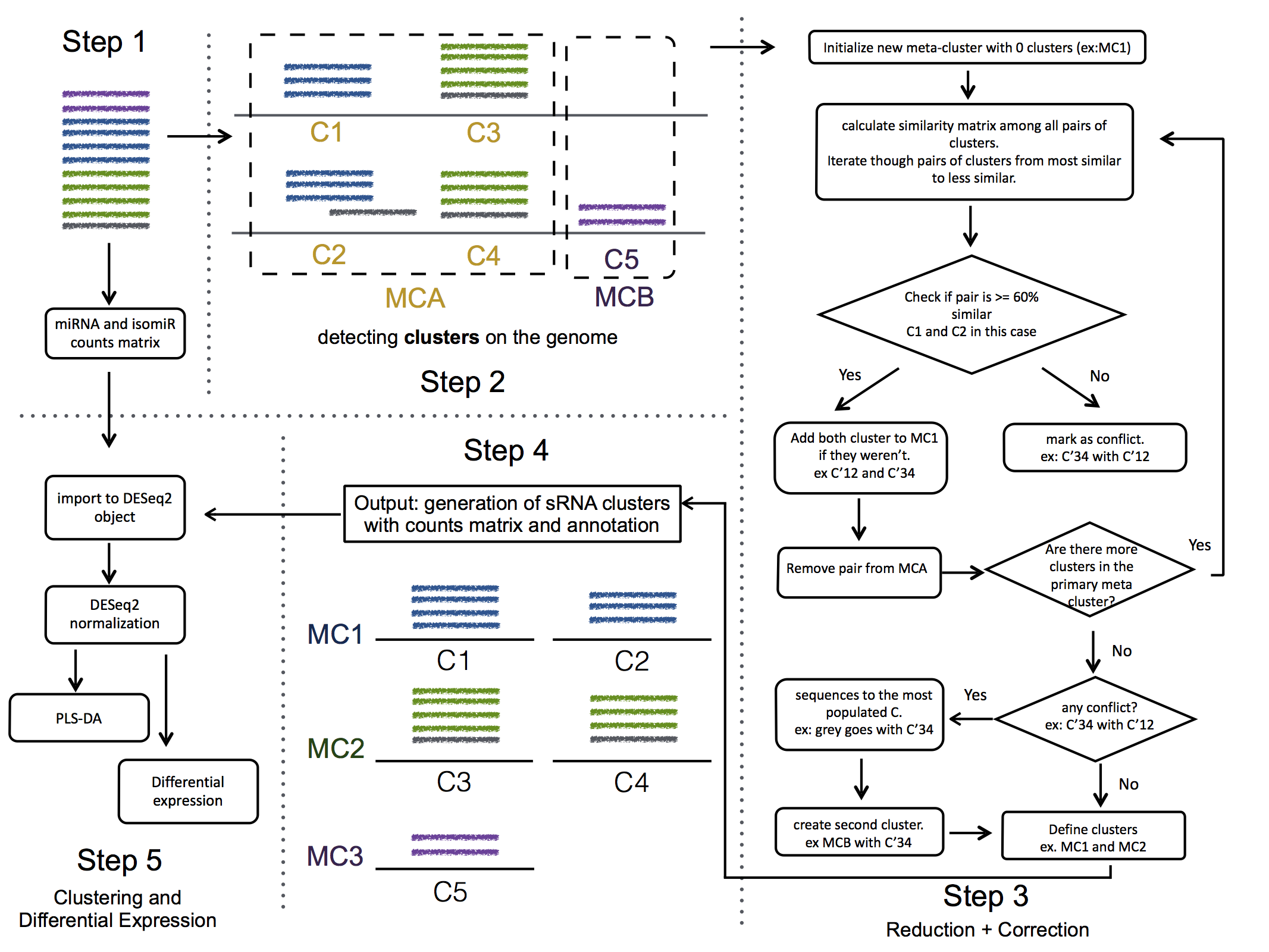
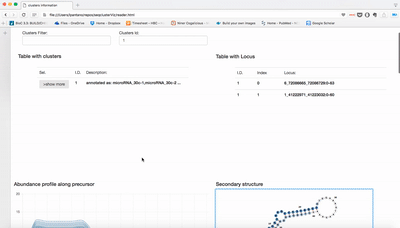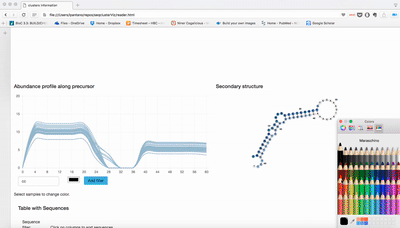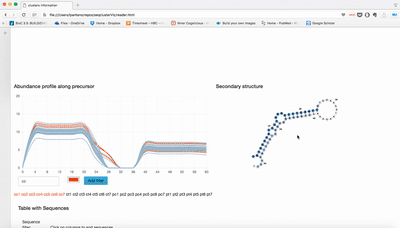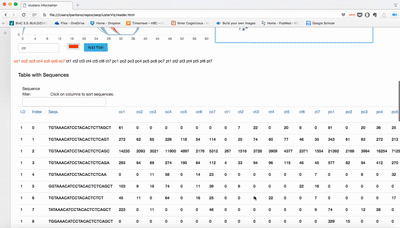
Interactive HTML Report¶
This will create html report using the following command assuming the output of seqcluster cluster is at res:
seqcluster report -j res/seqcluster.json -o report -r $GENONE_FASTA_PATH
where $GENOME_FASTA_PATH is the path to the genome fasta file used in the alignment.
Note: you can try our new visualization tool!
report/html/index.html: table with all clusters and the annotation with sorting optionreport/html/[0-9]/maps.html: summary of the cluster with expression profile, annotation, and all sequences insidereport/html/[0-9]/maps.fa: putative precursor
An example of the output is below:
Easy start with bcbio-nextgen.py¶
Note:If you already are using bcbio, visit bcbio to run the pipeline there.
To install the small RNA data:
bcbio_nextgen.py upgrade -u development --tools --datatarget smallrna
Options to run in a cluster
It uses ipython-cluster-helper to send jobs to nodes in the cluster
- –parallel should set to ipython
- –scheduler should be set to sge,lsf,slurm
- –num-jobs indicates how much jobs to launch. It will run samples independently. If you have 4 samples, and set this to 4, 4 jobs will be launch to the cluster
- –queue the queue to use
- –resources allows to set any special parameter for the cluster, such as, email in sge system: M=my@email.com
Read complete usability here: https://github.com/roryk/ipython-cluster-helper An examples in slurm system is:
--parallel ipython --scheduler slurm --num-jobs 4 --queue general
Output
- one folder for each analysys, and inside one per sample
- adapter: *clean.fastq is the file after adapter removal, *clean_trimmed.fastq is the collapse clean.fastq, *fragments.fastq is file without adapter, *short.fastq is file with reads < 16 nt.
- align: BAM file results from align trimmed.fastq
- mirbase: file with miRNA anotation and novel miRNA discovery with mirdeep2
- tRNA: analysis done with tdrmapper [citation needed]
- qc: *_fastqc.html is the fastqc results from the uncollapse fastq file
- seqcluster: is the result of running seqcluster. See its documentation for further information.
- report/srna-report.Rmd: template to create a quick html report with exploration and differential expression analysis. See example here
